")
Source : Paper Microbiology Indonesia, Vol. 4 , No. 3, Dec 2010, p 143-148
Written by : Juniastuti 1,4* , Eduardus Bimo Aksono 3 , Takako Utsumi 4,5 , Yoshihiko Yano 5 , Soetjipto 2,4 , Yoshitake Hayashi 5, Hak Hotta 5 , Fedik Abdul Rantam 5, Hernomo Ontoseno Kusumobroto 6 , Maria Inge Lusida 1,4
1Department of Microbiology, School of Medicine, Universitas Airlangga, Jalan Mayjen Prof Dr Moestopo 47, Surabaya 60131, Indonesia; 2Department of Biochemistry, School of Medicine, Universitas Airlangga, Jalan Mayjen Prof Dr Moestopo 47, Surabaya 60131, Indonesia; 3Institute of Tropical Disease, Universitas Airlangga, Jalan Mulyorejo, Surabaya 60115, Indonesia; 4 Indonesia-Japan Collaborative Research Center for Emerging and Re-emerging Infectious Diseases, Institute of Tropical Disease, Universitas Airlangga, Jalan Mulyorejo, Surabaya 60115, Indonesia; 5 Center for Infectious Diseases, Kobe University Graduate School of Medicine, 7-5-1 Kusunoki-cho, Kobe, Hyogo, 650-0017, Japan; 6 Department of Internal Medicine, Dr. Soetomo General Hospital, Jalan Mayjen Prof Dr Moestopo, Surabaya 60131, Indonesia.
Mutations of precore (A1896) and core promoter (T1762/A1764) of hepatitisBvirus can reduceHBeAgproduction. These mutations are frequently found in the late HBeAg seroconversion. However, it has been a controversy about the role played by precore and core promoter mutations in determining outcome of chronic hepatitis B. In the present study, the variability of precore and core promoter of hepatitisB virus were analyzed using PCR amplification and sequencing, according to the outcome (viral load and HBeAg/anti-HBe) in chronic hepatitis B patients in Surabaya. The study groups included 5 patients with uncomplicated chronic hepatitis B and 10 patients with chronic hepatitis B and liver cirrhosis in Dr. Soetomo Hospital, Surabaya. The control group included 6 blood donors obtained from Indonesia Red Cross, Surabaya. All groups were HBsAg positive. Precore mutation A1896 was predominant in all groups (60%-67% of each), together with precore variant T1858. As reported, precore variant T1858 is a prerequisite for precore A1896 and characteristic for viral genotype. Nevertheless, core promoter mutations T1762/A1764 were predominant only in LC patients (60%).All of these mutations were found mostly after HBeAg seroconversion (anti-HBe+). Of most samples with anti-HBe+, precore mutation was related with low viral load (<105 copies/mL), but core promoter mutations with high viral load (≤ 105 copies/mL). Precore mutation A1896 was predominant in all groups, but core promoter mutations T1762/A1764 were only predominant in LC patients. The precore mutation alone is possible not critical to indicate a poor outcome, the core promoter mutations must be considered also.
Key words: hepatitis B virus, precore mutations, core promoter mutations, chronic hepatitis B
*Corresponding author: Phone: 62-31-5030252 ext. 159, Fax: +62-31-5022472, E-mail: koeraisindewi@yahoo.co.id
Chronic hepatitis B virus (HBV) infection is a major health problem worldwide, especially in Indonesia which belongs to the moderate-to-high hepatitis B endemic region (Sastrosoewignjo et al. 1991; Khan 2004). HBV belongs to the Hepadnaviridae and replicates its DNA genome via a reverse transcription step. The high spontaneous error rate of the viral reverse transcriptase is responsible for viral genome evolution during the course of infection under the antiviral pressure of the host immune response or specific therapy (Summers and Mason 1982).
HBVinfection is associated with various courses of liver diseases ranging from asymptomatic carrier state to fulminant hepatitis. Recently, the clinical importance of HBV genome variability has been discovered. However the results of recent molecular studies are a matter of controversy, and these issues have yet to be resolved (Davidson et al. 2005).
In the natural course of chronic HBV infection, the early seroconversion from HBeAg to anti-HBe usually indicates a favorable outcome, because it is usually associated with the cessation of viral replication and non-progressive liver disease (Chen 1993; Chu 2000). In contrast, the late seroconversion of HBeAg after multiple bouts of reactivation and remission may accelerate the progression of chronic hepatitis to liver cirrhosis and, thus have a poor clinical outcome (Liaw et al 1987; Perrillo 2001). In clinical practice, the most frequently encountered variant form of chronic HBV infections is HBeAg-negative chronic hepatitis B associated with the replication of precore region mutants that terminate precore protein and HbeAg expression (Carman et al 1989; Rizetto and Ciancio 2008). The most frequently detected precore mutation is nucleotide transition at the codon 28 (A1896) which converts into a TAG stop codon (Okamoto et al 1990). Evidence has been proposed that the pattern of precore mutation is restricted by the secondary structure requirements of the epsilon encapsidation signals which are essential for its function and therefore by HBV genotypes. The presence of this mutation is dependent upon the nucleotide (C/T) of the precore at position 1858 (codon 15) (Lok et al 1994). Besides, the core promoter region also plays an important role in the viral life cycle, concerning the production of the precore mRNA and pregenomic RNA. HBV carrying the most common core promoter mutations, T1762 and A1764 displayed reduced levels of HBeAg synthesis and were associated with enhanced viral replication (Buckwold et al 1996). Previous reports have suggested that HBeAg mutations are associated with chronic hepatitis, liver cirrhosis and hepatocellular carcinoma (Rodriguez-Frias 1995). However, conflicting evidence suggests that HBeAg mutations are present in HBV carriers and in those individuals with mild forms ofHBVinfection or without liver disease (Davidson et al 2005).
The aim of this study was to analyze the precore and core promoter mutations in patients with uncomplicated chronic hepatitis B, patients with chronic hepatitis B and liver cirrhosis, and blood donors as controls, according to the outcome (viral load and HBeAg/anti-HBe status).
MATERIALS AND METHODS
Patients and controls. Three groups of individuals were included in this study. Group 1 included 5 patients with uncomplicated chronic hepatitis B, i.e. patients who were HBsAg positive more than 6 months without any clinical significance, confirmed by the medical records of the laboratory and clinical data. Group 2 included 10 patients with chronic hepatitis B and liver cirrhosis, i.e. patients who were HBsAg positive more than 6 months and had liver cirrhosis shown by the medical records of the laboratory and ultrasonography data. Group 1 and 2 were obtained from Dr. Soetomo General Hospital, Surabaya, Indonesia. Group 3 as the control group included 6 blood donors, i.e. individuals who were HBsAg positive and ALT normal, obtained from BloodTransfusion Unit – Indonesian Red Cross in Surabaya. Exclusion criteria included hepatitis C infection, HIV infection and use of antiviral therapy. The descent of each individual was documented, both maternally and paternally. All sera from each individual were stored at -20°C for further examinations. Ethical clearance of this study was obtained from the ethics committee of the Dr Soetomo General Hospital, in Surabaya, Indonesia. All participants signed an agreement to participate in this study.
HBeAg/anti-HBe Serology and Viral DNA Load. HBeAg and anti-HBe serologic assays were performed by microparticle EIA (Abbott). Quantitative determination of viralDNAin serum was performed by CobasAmplicorHBV monitor (Roche).
Viral DNA Extraction, PCR Amplification and Sequencing. The DNA of HBV was extracted from each serum sample using the High Pure Viral Nucleic Acid (Roche) following the manufacturer’s guidelines. The extracted DNAwas used as a template for the amplification of the respective gene regions. PCRs were performed with the High Fidelity PCR enzyme mix (Fermentas). The reactions contained 2.5 µL High Fidelity PCR buffer (with MgCl 2), 2.5 µL dNTP with a concentration 2 mM, 0.25 µL High Fidelity PCR enzyme mix, 10 µL DNA, and 0.5 µL of each primer with a concentration of 100 pmol L-1 , in a total reaction volume of 25 µL. The thermocycling condition included a 5-min denaturation step of 94°C , followed by 40 cycles of 1 min at 94°C , 1 min at 55°C , and 2 min at 72°C .
The partial core promoter gene and the precore gene were amplified in the first round using HBc1 (5′-TTA CATAAG AGGACTCTTGG-3′, nt 1650 to 1669) and HB9R (5′-GGA TAGAACTAGCAGGCAT-3′, nt 2654-2635) (Sugauchi et al 2001). If the first round PCR was negative, the second round PCR was performed using primers HBc1 and HBc2 (5′-TAAAGC CCAGTAAAG TTT CC-3′, nt 2494 to 2475) (Lusida et al. 2008).
To shed more light on the association between HBV genotype and precore mutation, part of the S gene was amplified in the first round using primers P7 (5′-GTG GTG GAC TTC TCT CAATTT TC-3′, nt 256 to 278) and P8 (5′-CGG TAW [A/T] AAA GGG ACT CAM [A/C] GAT-3′, nt 796 to 776). If the first round PCR was negative, the second round PCRwas performed using primers HBs1 (5′-CAAGGTATG TTG CCC GTT TG-3′, nt 455 to 474) and HBs2 (5′-AAA GCC CTG CGAACCACT GA-3′, nt 713 to 694) (Lusida et al 2008).
Nucleotide sequences of the amplified fragments were determined using the Big Dye Terminator v1.1 Cycle Sequencing Kit with an ABI Prism 310 (Applied Biosystems, USA).
Sequences Analyses for Detection of Precore and Core Promoter Mutations and Determination of HBV Genotypes. All analyses of the sequences data were performed with Genetyx-Mac version 9 (Software Development Co., Ltd., Tokyo, Japan). After aligning the sequences obtained from this study and from International DNAdatabases (DDBJ/EMBL/GenBank), the variability of the precore and core promoter sequences were analyzed. The HBV genotypes were determined based on the homologous percentage of >96% in the S gene sequences compared with HBV isolates from International DNA databases (DDBJ/EMBL/GenBank) (Magnius and Norder 1995; Arauz-Ruiz et al. 1997).
RESULTS
Atotal of 21 HBsAg-positive sera were obtained from 6 women and 15 men (aged 22 – 68 years). They were people of Java (85.7%), Flores (4.8%), Sulawesi (4.8%) and Aceh- Batak (4.8%) living in Surabaya. Of these, 13 (61.9%) were HBeAg-/anti-H Be+, 6 (28.6%) were HBeAg+/ anti-HBe-, and 2 (9.5%) were HBeAg-/anti-H Be-. The high HBVDNA levels, ≤105 copies mL-1 (13/21, 61.9%) were frequently found in patients with chronic hepatitis B and liver cirrhosis (7/10, 70%). The low HBV DNA levels were detected in 38.1% of the total sera, mostly found in controls (3/6, 50%).
HBV genotypes could be assigned to 15 HBV isolates. Based on part of the S gene, all of these isolates were classified intoHBVgenotypeB(Fig 1).
The most frequently detected mutation was G1896A (61.9%) in the precore region, and it was predominant in all groups (60-66.7% of each). The precore G1896A was the most prevalent in HBV isolates with genotype B (10/15, 66.7%) (Table 1). Besides, all isolates studied had precore variant T1858. The precore mutation, A1846T was detected in 33.3% isolates (7/21), mostly (5/7, 71.4%) associated with the virus carrying an A1896 or A1899. The other precore mutation G1899Awas detected in 23.8% isolates (5/21), and some of these were combined with A1896 (3/5, 60%) (Table 1, Fig 1). In the core promoter region, A1762T and G1764A were detected in some isolates (38% and 33.3%, respectively), and these mutations were predominant in patients with chronic hepatitisBand liver cirrhosis (60% and 50%, respectively) but were few in the other groups. The other core promoter mutations, T1753C (amino acid T127) and T1754G (amino acid M127) were detected in 33.3% isolates (7/21), and found around 30% – 40% in each group (Table 1, Fig 2).

Table 1 Variability of precore and core promoter in patients with uncomplicated chronic hepatitis B, patients with chronic hepatitis B and liver cirrhosis and controls
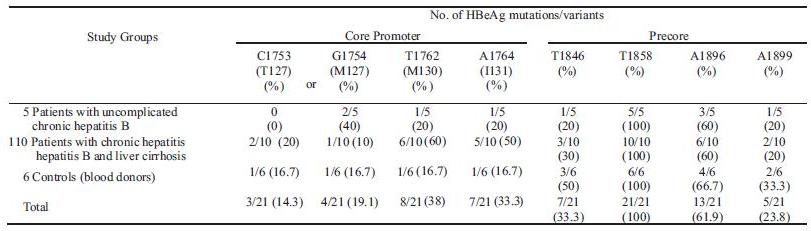

Fig 2 Nucleotide sequences of the partial core promoter region of HBV isolates from patients with uncomplicated chronic hepatitis B (U), patients with chronic hepatitis B and liver cirrhosis (S) and controls (P). The two upper sequences were downloaded from International DNA databases (DDBJ/EMBL/GenBank).
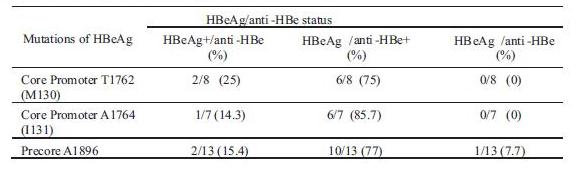
The most prevalent precore mutation, A1896 was frequently detected in HBeAg-/anti-HBe+ status (77%). The core promoter mutations, T1762 and A1764 were also predominant in sera with HBAg-/anti-HBe+ (75% and 85.7%, respectively). These precore and core promoter mutations were detected few in the other HBeAg/anti-Hbe status (0-25%) (Table 2).
Table 2 Mutations of precore and core promoter according to the HBeAg/anti-HBe status
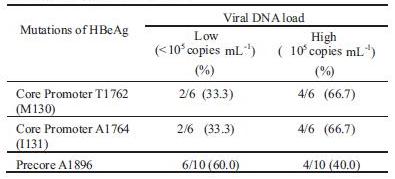
Of most sera with antiHBe+, precore A1896 was mostly related with low HBV DNA levels (60%). On the contrary, core promoter T1762 and A1764 were frequently related with highHBVDNAlevels (66.7%) (Table 3)
Table 3 Mutations of precore and core promoter according to the viral DNA load in samples with anti-HBe+
DISCUSSION
A nucleotide transition which converts into a TAG stop codon mutation (A1896) of the precore region develops during viral replication and displacement of the wild type by mutant can take several years (Okamoto et al 1990; Akarka et al 1994). This mutation prevents the translation of the precore protein and completely abolishes the production of HBeAg (Kao 2002). Although HBeAg is not required for viral replication or infectivity, its exact function is not known (Kramvis and Kew 2005). HBeAg is believed to be an important humoral and cellular immune target, this loss of HBeAg production may represent escape mutation, thus contributing to viral persistence (Carman et al 1993). Detailed studies showed a significant link between precore mutation with remission of liver disease (Chan 1999), but others found a high prevalence of precore mutation in patient with severe liver disease (Chisari 1997; Friedt et al 1999; Kao 2000).
The result of this study showed that the precore A1896 was predominant not only in patients with chronic hepatitis B and liver cirrhosis, but also in patients with uncomplicated chronic hepatitis B and controls (60-66.7% of each) (Table 1). It has been considered that a contributing factor in precore mutation frequency is viral genotype (Kramvis and Kew 2005). The precore A1896 is found only in patients infected with HBV genotypes B, D, E, and in minority only of the genotypes C and F strains that bear a variant T at nt 1858 (Hadziyannis 1995; Kramvis and Kew 2005). The precore A1896 and T1858 tighten the stem structure by making aT-Apair (Lok et al 1994), although T1858 can also make a wobble pairing with G1896 (Kao 2002). The HBV isolates with certain genotypes which had precore T1858 are more frequent in geographic regions such as Asia and the Mediterranean basin (Rodriguez-Frias et al 1995; Lindh et al 1997) related with the ethnic origin of the individual carriers of the HBV (Davidson et al 2005). As previous studies (Lusida et al 2003; Mulyanto et al 2009), this study showed that HBV genotype B was predominant in patients with HBV infection (mostly Javanese) in Surabaya, and it agreed with the predominance of precore A1896 in all groups. However, the association between precore A1896 and other genotypes can not be explained in this study. Considering clinical outcomes, this study supports some previous studies (Zarski et al 1994), that precore A1896 alone may have no direct pathogenic role, especially in HBV isolates with some genotypes having precore T1858.
The precore A1896 combined with other precore mutations was detected in some HBV isolates. As much as 7 isolates (33.3%) had an A to T mutation at nt 1846 in the precore region, mostly associated with the presence of 1896 or 1899 mutation (Table 1, Fig 1). The double precore mutations at nt 1896 and nt 1899 was detected in 3 isolates (14.3%) (Fig 1). As proposed, precore A1896 and T1858 together with precore A1899 and T1855 could enhance the stability of the lower stem by replacing T-G pair with more stable T-A pair (Lok et al 1994). Besides, although the mutation at nt 1846 will destroy base pairing at nt 1896 or nt 1899, the combined mutation at 1896 or 1899 may stabilize stem loop (Cho et al 1999). Some of these combined mutations at nt 1846-nt 1896/1899 and at nt 1896-nt 1899 were found in sera with low HBV DNA levels (60% and 66.6%, respectively) and with high HBV DNA levels (40% and 33.3%, respectively) (data not shown). Further studies are necessary to confirm the significance of these mutations on viral replication.
Mutations in the core promoter that is part of the X region can also prevent HBeAg production by selectively down-regulating the transcription of the precore mRNA. The most common mutations involved A to T change at nt 1762 (amino acid K130M) and G to A change at nt 1764 (amino acid V131I) (Okamoto et al 1994). These mutations were mostly detected in patients with chronic hepatitis B and liver cirrhosis (50-60%), and found few in other groups (16.7-20%) (Table 1). Utama et al (2009) also reported that the double mutations were only found in 1.96% blood donors in Makasar, Indonesia. As Kramvis and Kew reported (2005), the core promoter mutations, T1762 andA1764 were found to be significantly associated with more severe liver disease (liver cirrhosis with or without hepatocellular carcinoma). It has been proposed that core promoter, T1762/A1764 may enhance HBV virulence by increasing host immune response, increasing viral replication, or altering the coding region for the X (Kidd-Ljunggren et al 1995; Buckwold et al 1996; Li et al 1999; Hunt et al 2000). By diminishing circulating HBeAg, core promoter mutants may augment the host immune response to HBV-infected hepatocytes, hence increasing hepatocytes apoptosis and regeneration, which leads to liver injury (Hunt et al 2000). Interestingly, most HBV isolates with core promoter, T1762/A1764 were not accompanied with precore A1896 (5/8, 62.5%), especially in patients with chronic hepatitis B and liver cirrhosis (Fig 1-2). It is still unclear the importance of the precore A1896 in pathogenesis of HBV with core promoter T1762/A1764.
The other core promoter mutations, C1753 (amino acid T127) and G1754 (amino acid M127) were detected in 7 HBV isolates (33.3%) (Table 1, Fig 2). The T127 mutation was recently described in Japanese patients with fulminant and chronic hepatitis (Kreutz 2002), and it was shown that this mutation affecting the crucial domain for transactivation suppress the transcription of both precore and core mRNA. In this study, the T127 mutation was detected around 0 – 20% in each group (Table 1). It needs further investigation to confirm its role.
The most frequently encountered variant form of chronic HBV infections is HBeAg negative chronic hepatitis B due to replication of naturally occurring HBV variants with nucleotide substitutions in the precore and/or core promoter regions of the genome and represents a later phase of chronic HBV infection. HBV can mutate into an HBeAg negative phenotype to evade the anti-HBe immune pressure that develops sooner or later in hosts. HBeAg-negative mutants frequently become the predominant virus population in chronic HBsAg carriers, possibly indicating a selection advantage against the wild type (Okamoto et al 1990; Okamoto et al 1994). The major mutations focused in this study, precoreA1896 and core promoter T1762/A1764 were mostly found in sera with antiHBe+ (Table 2). Of most sera with anti-HBe+, precore A1896 was related with low viral load (<10 copies mL ), but core promoter T1762/A1764 with high viral load ( 10 copies mL ) (Table 3). Lin et al (2007) also reported that patients with core promoter T1762/A1764 mutant had significantly higher serum HBV DNA levels than those with core promoter A1762/G1764 wild type strain, regardless of precore 1896 status. It appears that the mutations of core promoter, T1762/A1764 were related with severe liver disease after HbeAg seroconversion not as precore A1896, and it might not to worry about the high prevalence of precore A1896 in Surabaya.
Interestingly, two sera obtained from one patient with uncomplicated chronic hepatitis B (7RS) and one patient with chronic hepatitis B and liver cirrhosis (17RS), had precore mutation A1896 but HBeAg did not disappear (data not shown). It might be due to presence of a mixed infection of the mutant and wild type viruses. The presence of wild type virus is needed by some of the mutants for infection of hepatocytes. It appears that HBV exists as a quasi species of wild type and mutant clones even in the HBeAg positive phase (Davidson 2005). The INNO-LIPAassay will be able to pick up the mixed infection (Abbas 2006). A heterogenous virus population circulating in patients with chronic HBV infection must be considered to determine the outcome.
Overall, it could be concluded that precore mutation A1896 was predominant in all groups, but core promoter mutations T1762/A1764 were only predominant in patients with chronic hepatitis B and liver cirrhosis. The precore mutation alone is possibly not critical to indicate a poor outcome, core promoter mutations must be considered also. Further studies with a larger population are needed to investigate which type of specific mutations or which combined mutations is associated with severe iver disease and thus might play a role in pathogenesis.
ACKNOWLEDGEMENTS
The authors are grateful to Slamet Riyadi, Nur Achmad Tjipto, and Nasronudin for their cooperations; also Mochamad Amin and Koen Poedijati for their technical assistance. We also deeply appreciate the patients at Dr Soetomo General Hospital, Surabaya and blood donors at the Blood Transfusion Unit-Indonesia Red Cross, Surabaya, who had given their blood in this study. This study was supported by a grant from the Directorate General of High Education, Department of National Education, Indonesia, and also by the Program of Founding Research Centers for Emerging and Reemerging Infectious Diseases, the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.
REFERENCES
- Abbas Z, Muzaffar R, Siddiqui A, Naqvi SAA, Rizvi SAH. 2006. Genetic variability in the precore and core promoter regions of hepatitis B virus strains in Karachi. BMC Gastroenterology 6:20, doi: 10.1186/1471-230X-6-20.
- Akarka US, Greene S, Lok ASF. 1994. Detection of precore hepatitis B virus mutants in asymptomatic HBsAg-positive family members. Hepatology 19:1366-70.
- Arauz-Ruiz P, Norder H, Visoná KA, Magnius LO. 1997. Molecular epidemiology of hepatitis B virus in Central America reflected in the genetic variability of the small S gene. J Infect Dis 176:851-8.
- Buckwold VE, Xu ZC, Chen M,Yen TS, Ou JH. 1996. Effects of a naturally occurring mutation in the hepatitis B virus basal core promoter on precore gene expression and viral replication. JVirol 70: 5845-51.
- Carman W, Hadziyannis S, McGarvey MJ, Jacyna M, Karayiannis P, Makris A, Thomas HC. 1989. Mutation preventing formation of hepatitis B e antigen in patients with chronic hepatitis B infection. Lancet 2:588–91.
- Carman WF, Thomas HC, Domingo E. 1993. Genetic variation in viruses: clinical relevance toHBV. Lancet 341:349-53.
- Chan HL, Hussain M, Lok AS. 1999. Different hepatitis B virus genotypes are associated with different mutations in the core promoter and precore regions during hepatitis B e antigen seroconversion. Hepatology 29:976-84.
- Chen DS. 1993. From hepatitis to hepatoma: lessons from type B viral hepatitis. Science 262:369-70.
- Chisari FV. 1997. Perspective series: host/pathogen interactions. J Clin Invest 99:1472-7.
- Cho SW, Shin YJ, Hahm KB, Jin JH, Kim YS, Kim JH, Kim HJ. 1999. Analysis of the precore and core promoter DNA sequence in liver tissues from patients with hepatocellular carcinoma. J Korean Med Sci 14:424-430.
- Chu CM. 2000. Natural history of chronic hepatitis B virus infection in adults with emphasis on the occurrence of cirrhosis and hepatocellular carcinoma. J Gastroenterol Hepatol 15:E25-30.
- Davidson F, Lycett C, Sablon E, Petrik J, Dow BC. 2005. Hepatitis B virus genotypes and precore mutations in Scottish blood donors. Vox Sang 88:87-92.
- Friedt M, Gerner P, Lausch E, Trubel H, Zabel B,Wirth S. 1999. Mutations in the basic core promoter and the precore region of the hepatitisBvirus and their selection in children with fulminant and chronic hepatitis B. Hepatology 29:1252-8.
- Hadziyannis SJ. 1995. Hepatitis e antigen negative chronic hepatitis B: from clinical recognition to pathogenesis and treatment. Viral Hepat Rev 1:17-36.
- Hunt CM, McGill JM, Allen MI, Condreay LD. 2000. Clinical relevance of hepatitisBvirus mutations. Hepatology 31:1037-1044.
- Kao JH. 2002. Hepatitis B viral genotypes: clinical relevance and molecular characteristics. J Gastroenterol Hepatol 17:643-50.
- Kao JH, Chen PJ, Lai MY, Chen DS. 2000. Clinical and virological aspects of blood donors infected with hepatitis B virus genotypes B and C. J Clin Microbiol 40:22-5.
- Khan M, Dong JJ,Acharya SK, DhagwahdorjY,Abbas Z, JafriW, Mulyono DH, Tozun N, Sarin SK. 2004. Hepatology issues in Asia: perspective from regional leaders. J Gastroenterol Hepatol 19:419-30.
- Kidd-Ljunggren K, ObergM, KiddAH. 1995. The hepatitis B virusXgene: Analysis of functional domain variation and gene phylogeny using multiple sequence. J GenVirol 76:2119-30.
- Kramvis A, Kew MC. 2005. Relationship of genotypes of hepatitis B virus to mutations, disease progression and response to antiviral therapy. J Viral Hepat 12:456-64.
- Kreutz C. 2002. Molecular, immunological and clinical properties of mutated hepatitisBviruses. J Cell MolMed6:113-43.
- Li J, Buckwold VE, Hon MW, Ou JH. 1999. Mechanism of suppression of hepatitis B virus precore RNA transcription by a frequent double mutation. JVirol 73:1239-44.
- Liaw YF, Tai DI, Chu CM, Pao CC, Chen TJ. 1987. Acute exacerbation in chronic type B hepatitis: Comparison between HBeAg and antibodypositive patients. Hepatology 7:20-3.
- Lindh MA,Anderson AS, Gusdal A. 1997. Genotypes, nt1858 variants, and geographic origin of hepatitisB virus: Large-scale analysis using a new genotyping method. J Infect Dis 175:1285-93.
- Lok ASF, Akarca U, Greene S. 1994. Mutations in the precore region of hepatitis B virus serve to enhance the stability of the secondary structure of the pregenome encapsidation signal. Proc Natl Acad Sci USA91:4077-81.
- Lusida MI, Nugrahaputra VE, Soetjipto, Handajani R, Nagano-Fujii M, Sasayama M, Utsumi T, Hotta H. 2008. Novel subgenotypes of hepatitis B virus genotypes C and D in Papua, Indonesia. J Clin Microbiol 46:2160-6.
- Lusida MI, Surayah, Sakugawa H, Nagano-Fujii M, Soetjipto, Mulyanto, Handajani R, Boediwarsono, Setiawan PB, Nidom CA, Ohgimoto S, Hotta H. 2003. Genotype and subtype analyses of hepatitis B virus (HBV) and possible co-infection of HBV and hepatitis C virus (HCV) or hepatitis D virus (HDV) in blood donors, patients with chronic liver disease and patients on hemodialysis in Surabaya, Indonesia. Microbiol Immunol 47:969-75.
- Magnius LO, Norder H. 1995. Subtypes, genotypes and molecular epidemiology of the hepatitis B virus as reflected by sequence variability of the S-gene. Intervirology 38:24-34.
- Mulyanto, Depamede SN, Surayah K, Tsuda F, Ichiyama K, Takahashi M, Okamoto H. 2009. A nationwide molecular epidemiological study on hepatitis B virus in Indonesia: identification of two novel subgenotypes, B8 and C7.ArchVirol 154:1047-59.
- Okamoto H, Tsuda F,AkahaneY, SugaiY,Yoshiba M, Moriyama K, Tanaka T, MiyakawaY, Mayumi M. 1994. Hepatitis B virus with the mutations in the core promoter for an e antigen-negative phenotype in carriers with antibody to e antigen. JVirol 68:8102-10.
- Okamoto H, Yotsumoto S, Akahane Y, Yamanaka T, Miyazaki Y, Sugai Y, Tsuda F, Tanaka T, MiyakawaY, Mayumi M. 1990. Hepatitis B viruses with precore region defects prevail in persistently infected hosts along with seroconversion to the antibody against e antigen. J Virol 64:1298-303.
- Perrillo RP. 2001. Acute flares in chronic hepatitis B: The natural and unnatural history of an immunologically mediated liver disease. Gastroenterology 120:1009-22.
- Rizetto M, Ciancio A. 2008. Chronic HBV-related liver disease. Mol AspectsMed29:72-84.
- Rodriguez-Frias F, Buti M, Jardi R, Cotrina M, Viladomiu L, Esteban R, Guardia J. 1995. Hepatitis B virus infection: precore mutants and its relation to viral genotypes and core mutations. Hepatology 22: 1641-7.
- Sastrosoewignjo RI, Sandjaja B, Okamoto H. 1991. Molecular epidemiology of hepatitisBvirus in Indonesia. J Gastroenterol Hepatol 6:491-8.
- Sugauchi F, Mizokami M, Orito E, Ohno T, Kato H, Suzuki S, Kimura Y, Ueda R, Butterworth LA, Cooksley WGE. 2001. A novel variant genotype C of hepatitis B virus identified in isolates from Australian Aborigines: complete genome and phylogenetic relatedness. J Gen Virol 82:883-92.
- Summers J, Mason WS. 1982. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29:403-15.
- Utama A, Octavia TI, Dhenni R, Miskad UA,Yusuf I, Tai S. 2009. Hepatitis B virus genotypes/subgenotypes in voluntary blood donors in Makasar, South Sulawesi,Indonesia.Virol J6:128, doi:10.1186/1743-422X-6-128.
- Zarski JP, Marcellin P, Cohard M, Lutz JM, Bouche C, Rais A, et al 1994. Comparison of anti-HBe-positive and HBe-antigen-positive chronic hepatitisBin France. J Hepatol 20:636-40.